Pepmatch
The Pepmatch tool scans a set of peptides against a large database of proteins for sequence identity at or below a specified number of mismatches.
Methods
Pepmatch uses an efficient, deterministic algorithm to perform the scanning process.
Parameter Selection
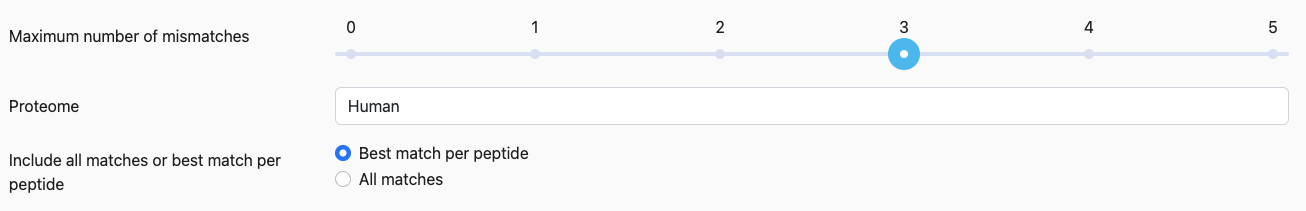
Maximum number of mismatches
When scanning for hits in the protein database, Pepmatch will return those that have this number of mismatches or less.
Proteome
The database of proteins to search against. All included proteomes were obtained from Uniprot/Swiss-Prot.
Available proteomes include:
human
mouse
cow
dog
horse
pig
rabbit
rat
Include all matches or the best match per peptide
Best match per peptide: Returns only one match per query peptide.
All matches: Returns all matches at or below the mismatch threshold for the query peptide.
Results
The pepmatch output will look similar to the table below:
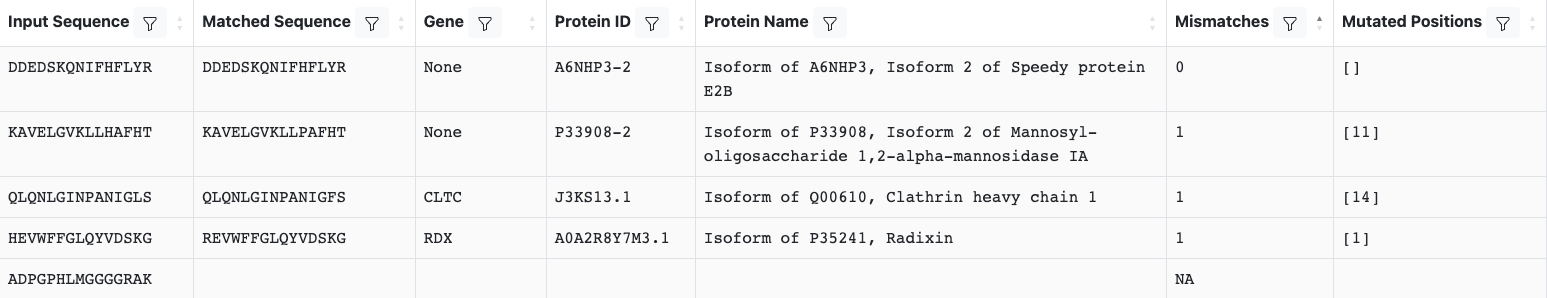
A row will be returned for every input peptide, regardless of whether a match was found. Peptides that do not match anything in the selected database will have mostly empty fields, except for ‘NA’ in the ‘Mismatches’ fields.
Some of the columns in the table include:
Input Sequence
Input peptide (query) sequence
Matched Sequence
Matched peptide sequence from the proteome database.
Gene
The gene name associated with the peptide hit.
Protein ID
The Uniprot/Swissprot accession associated with the peptide hit.
Protein Name
The name of the protein corresponds to the UniProt accession.
Mismatches
The number of mismatches between the query and matched peptides
Mutated Positions
Indexes of the positions that differ between the query and matched peptides.
Note
Although Pepmatch is an efficient algorithm, the current implementation on the NG Tools site is suboptimal and can take excessive time. This will be addressed in the next release.